How to Use DNNESCAN
In this page:
BIOLOGICAL QUESTIONS ANSWERED BY DNNESCAN
DNNESCAN helps the researchers to answer several biological questions on the transcriptional regulation of the yeast. For instance,
- What are the potential binding sites of a particular TF in a set of DNA sequences?
- Which TFs regulate a particular gene?
- Which genes are regulated by a specific TF?
PROGRAM ARGUMENTS
| Argument |
Required |
Description |
| -op or -operation |
Yes |
Search the DNA sequences for potential TFBSs [scan, predict] or build DNNESCAN model from a set of sequences [build, learn]. The "build" operation is not available for normal users. |
| -i or -input |
Yes |
It contains the genomic regions to be searched by DNNESCAN. |
| -f or -format |
No |
It determines the format of the input file [fasta, positions, genes]. It can be either in FASTA format, contain genomic positions or have locus tags of genes with the length of upstream region. If the length of upstream region is not provided in the "genes" format, the whole intergenic region (IGR) upstream the gene is scanned. Default is FASTA format files. |
| -p or -tf |
Yes |
DNNEScan currently supports the following TF proteins from the yeast genome:
ABF1, CBF1, CIN5, DIG1, FHL1, FKH1, GCN4, HAP1, MBP1,
NDD1, NRG1, PHD1, RAP1, REB1, SKN7, SOK2, STE12, SUT1,
SWI4, SWI5, SWI6, UME6, ALL. Users can use any of these gene symbols as a value for this argument. If ALL is selected, the DNA sequences are searched using all DNNESCAN models. More TFBSs recognition models will be available later. |
| -m -model |
No |
The DNNESCAN model file if users have a model which is not supported by the current release. |
| -o or -output |
No |
The output file name if the output format is set to "plain" or the output directory if output format is set to "html". If nothing is provided, an output file/directory will be created in the current directory. |
| -of or -outputformat |
No |
The format of DNNESCAN's report [plain, html]. Default is "html". |
| -overlap or -overlapping |
No |
Remove overlapping of TFBSs at different levels [tf, all, none]. "tf" removes overlapping between TFBSs of the same TF protein only. "all" removes overlapping between TFBSs of all TF proteins. "none" disables the overlapping procedure. Default is "all". When overlapping occurs, the TFBSs with the highest DNNE prediction score are retained. |
USAGE
To run DNNESCAN,
- Download DNNESCAN from the download page and unzip the file.
- Open the CMD in Windows or the Terminal in Linux.
- Change your current directory to DNNESCAN's directory.
- type in the command line
java -jar isdmotif_dnnescan.jar -< argument1 > <argument1 value> -< argument2 > <argument2 value> ...
Sample Runs
-
To look for potential binding sites for ABF1 in different file formats, type in the command line
java -jar isdmotif_dnnescan.jar -op scan -p abf1 -i test_abf1.fasta -f fasta -o predicted_sites_fasta.out
java -jar isdmotif_dnnescan.jar -op scan -p abf1 -i test_abf1.fasta -f fasta -o predicted_sites_fasta -of html
java -jar isdmotif_dnnescan.jar -op scan -p abf1 -i test_abf1.pos -f positions -o predicted_sites_positions.out
java -jar isdmotif_dnnescan.jar -op scan -p abf1 -i test_abf1.pos -f positions -o predicted_sites_positions -of html
java -jar isdmotif_dnnescan.jar -op scan -p abf1 -i test_abf1.orf -f genes -o predicted_sites_genes.out
java -jar isdmotif_dnnescan.jar -op scan -p abf1 -i test_abf1.orf -f genes -o predicted_sites_genes -of html
-
You can use DNNESCAN to search for occurrences of multiple TFs. For example, to look for potential binding sites for ABF1 and DIG1, type in the command line
java -jar isdmotif_dnnescan.jar -op scan -p abf1,dig1 -i test_abf1.fasta
-
To look for potential TFBSs for all supported TFs, type
java -jar isdmotif_dnnescan.jar -op scan -p all -i test_abf1.fasta
-
You can use DNNESCAN to search DNA sequences using a DNNESCAN model that is not supported by the current release. For example,
java -jar isdmotif_dnnescan.jar -op scan -m DNNESCAN_ABF1.model -i test_abf1.fasta -of html -overlap all
INPUT FILES
DNNESCAN can accept two input files:
- The DNA sequence file: it is a text file that contains the genomic sequences to be searched by DNNESCAN. The file can be in one of the following formats: fasta, positions or genes. Next, some details are provided for each format.
- FASTA format: it is a standard fasta file with some specifications. The header of each sequence should have the following format:
> sequence ID, chromosome number (as Roman Numeral), start -end (absolute positions in the chromosome)
For example,
>sequence BSDA, II, 257976-259868
GTTCGATCCCTACACCGACGTACGATGCAACTGTGTGGATGTGACGAGCTTCATTTATACGCTTCGCGCGCCGGAC
CGGCCTCCGCAAGGCGCAGCAGTGCACAAGCAAATGACAATTAACCACCGTGTATTCGTTATAGCATCAGGCAGTT
TAAGTCGGGACAATAGGGGCCGCAATACACAGTTTACCGCATCTTGACCTAACTGACAAACTGCCATGGACGACTA
GCCATGCCATTGGCTCTTAGACAGCCCGATACAGTGATTATGAAAGGTTTGCGGGGCATGGCTACGACTTGCTCAG
CTACGTGCGAGGGCAGAAACTTTTCCGCATTTGTATGTTCACCTATCTACTACCCATGCCCGGAGATTATGTAGGT
TGTGAGATGCGGGAGAGGTTCTCGATCTTCCCGTGGGACGTCAACCTTTCCCTTGATAAAGCATTCCGCTCGGGTA
TGGCAGTAAGTACGCCTTCTGAATTGTGCTAACCTTCATCCTTATCAAAGCTTGCTGCCAATGATTAGGATTATTG
CCTTGCGACAGACTTCCTACTCACACTCGCTCACATTGAGCTACTCGATGGGCCATCAGCTTGACCCGCTCTGTAG
GGTCGCGATTACGTGAGTTAGGGCTCCGGACTGCGCTGTATAGTCGAATCTGATCTCGCCCCCACAACTGCAAACC
CCAACTTATTTAGATAACATGATTAGCCGAAGTTGCACGGGGTGCCCACCGTGGACTCCTCCCCGGGTGTCGCTCC
TTCATCTGACAATATGCAGCCGCTACCACCATCGATTAATACAACGAACGGTGATGTTGTCATAGATTCGGCACAT
TTCCCTTGTAGGTGTGAAATCACTTAGCTTCGCGCCGAAGTCTTATGGCAAAACCGATGGACTATGTTTCGGGTAG
CACCAGAAGTCTATAGCACGTGCATCCCAACGTGGCGTGCGTACACCTTAATCACCGCTTCATGCTAAGGTCCTGG
CTGCATGCTATG
If the sequence's start and end positions are not provided in the sequence header, the sequence is aligned against the yeast genome to identify its location.
- POSITIONS format: it is a three-column text file in which each genomic sequence is encoded in one line: the first column represents the sequence chromosome and the second and third columns represent the absolute start and end positions of the sequence, respectively.
For example,
II 257976-259868
II 534818-535259
I 45023-45898
III 258655-258882
XIV 405565-406357
- GENES format: it is a two column text file that contains the target genes whose upstream promoters will be scanned. The gene locus is used to represent genes in the first column. The second column contains the number of base pairs upstream the gene.If the length of upstream region is not provided in the "genes" format, the whole intergenic region (IGR) upstream the gene is scanned.
For example,
YBR012W-A 2000
YBR146W
YAL053W
YCR082W 1000
YNL118C
- The DNNESCAN model file: it is a binary file generated by DNNESCAN developers only and can be provided to users to search for putative TFBSs.
OUTPUT FILES
DNNESCAN generates search reports in two formats:
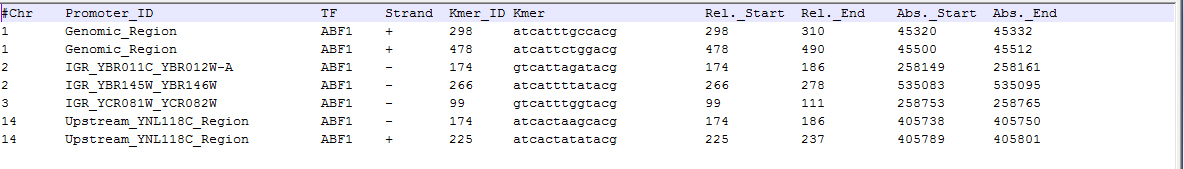
- Plain format: the discovered TFBSs are exported into a text file of tabular format. The file consists of ten columns as follows:
- Chromosome ID: the chromosome number of the DNA sequence.
- Promoter ID: an identifier assigned to the DNA sequence.
- TF Protein: the TF protein bound to this binding site.
- Strand: the TFBS is located on the forward (+) or reverse (-) strand.
- K-mer ID: an identifier is assigned to each discovered TFBS.
- K-mer: the DNA sequence of the putative TFBS.
- Relative Start: the relative start position of the TFBS in the parent DNA sequence (one-based).
- Relative End: the relative end position of the TFBS in the parent DNA sequence (one-based)
- Absolute start: the absolute start position of the TFBS in the parent chromosome (one-based)
- Absolute End: the absolute end position of the TFBS in the parent chromosome (one-based)
For example,
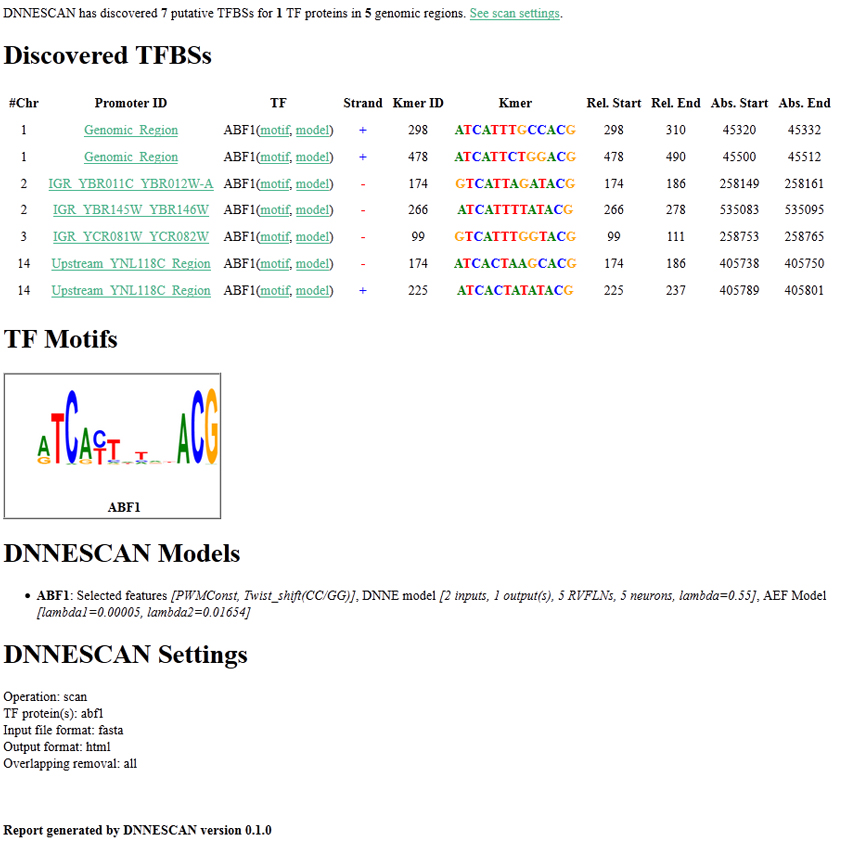
- HTML format: DNNESCAN creates an interactive HTML report that shows the TFBS search results and the parameter values of the used DNNESCAN models. The report is organized into HTML files. It has an index page and additional pages, one for each genomic sequence. To see the report, open the "index.html" page in the output directory. The index page consists of three sections:
- Discovered TFBSs: table of the identified binding sites in all input DNA sequences. It is similar to the table generated when the "plain" format is selected.
- TF Motifs: the sequence logos of the motifs of all the used TFs.
- DNNESCAN Models: the parameter values of the DNNESCAN models for each TF. It shows the selected features, the DNNE model parameters and the adaptive ellipsoidal filter (AEF) model parameters.
- Scan Settings: the program settings when the report is generated.
For example, a typical HTLM report looks like
If the user clicks on the promoter id of a sequence in the table, the report navigates to a new page that shows that sequence with all the discovered TFBSs highlighted with yellow. If there is an overlapping between neighbouring TFBSs on the same strand, the light green colour is used alternatively with the yellow. For example,

If you hover on the discovered binding sites, additional information is displayed as a tip.
See the case studies to see more examples on the HTML reports.